
Découvert le 25 novembre dernier et déclaré « préoccupant » par l’OMS le lendemain, le variant Omicron (B.1.1.529) est suivi de près par de nombreuses équipes. Chercheurs au sein de l’unité « Maladies infectieuses et vecteurs : Écologie, Génétique, Évolution et Contrôle » (Université de Montpellier, CNRS, IRD), Mircea Sofonea, maître de conférences, et Samuel Alizon, directeur de recherche, spécialistes de l’épidémiologie et de l’évolution des maladies infectieuses, reviennent sur la dynamique des variants. Prépondérance de Delta, particularités d’Omicron… Explications en 10 points clés par les deux spécialistes.
The Conversation : Pourquoi Delta a-t-il écrasé tous les autres variants du SARS-CoV-2 pendant des mois ?
Samuel Alizon : Le variant Delta est assez « monstrueux ». Cela se voit par exemple au niveau des estimations du nombre de reproduction de base, R₀ (nombre moyen d’infections que cause une personne infectée, dans une population donnée). Notre équipe l’a évalué aux alentours de 3 dans un rapport de mars 2020 pour les lignées ancestrales, en France. Pour le variant Alpha, le R₀ était entre 4 et 5 ce qui explique son invasion rapide début 2021. Pour Delta, les estimations sont entre 6 et 8.
On pourrait presque parler d’un avantage « qualitatif » sur les autres variants, comme le signalent les études de terrain : si contrôler la propagation des lignées ancestrales revenait à stopper la propagation d’une grippe pandémique (R₀ < 3), avec Delta cela s’apparente plus à contrôler un virus comme la rubéole (R₀ > 5). Ce choc est particulièrement violent pour les populations peu vaccinées ou immunisées, comme on l’a vu cet été aux Antilles ou plus récemment en Europe de l’Est.
Mircea T. Sofonea : Parmi les virus respiratoires humains, seuls ceux des oreillons, de la varicelle et de la rougeole sont plus contagieux, avec des R₀ souvent estimés à plus de 10. Et plus un virus se propage vite, plus un autre variant qui n’est qu’un peu plus contagieux tardera à émerger.
C’est un résultat de biologie de l’évolution qu’illustre le modèle géométrique de Fisher qui date des années 1930. Schématiquement, il revient à considérer chaque mutation comme un déplacement aléatoire de proche en proche sur un paysage dont le relief représente la capacité du virus à se propager dans la population humaine. Par sélection naturelle, seuls les déplacements correspondant à une ascension sont conservés. Le modèle de Fisher suggère que cette ascension se fait de plus en plus lentement, car la probabilité qu’un déplacement aléatoire tombe sur un point plus élevé diminue avec la proximité du sommet du paysage, l’optimum adaptatif.
De fait, depuis juillet 2021, le nombre de variants n’a pas explosé et nous avons plutôt assisté à la diversification de la lignée du variant Delta en une centaine sous-lignées – dont certaines sont surveillées plus étroitement du fait des mutations d’intérêt qu’elles portent.
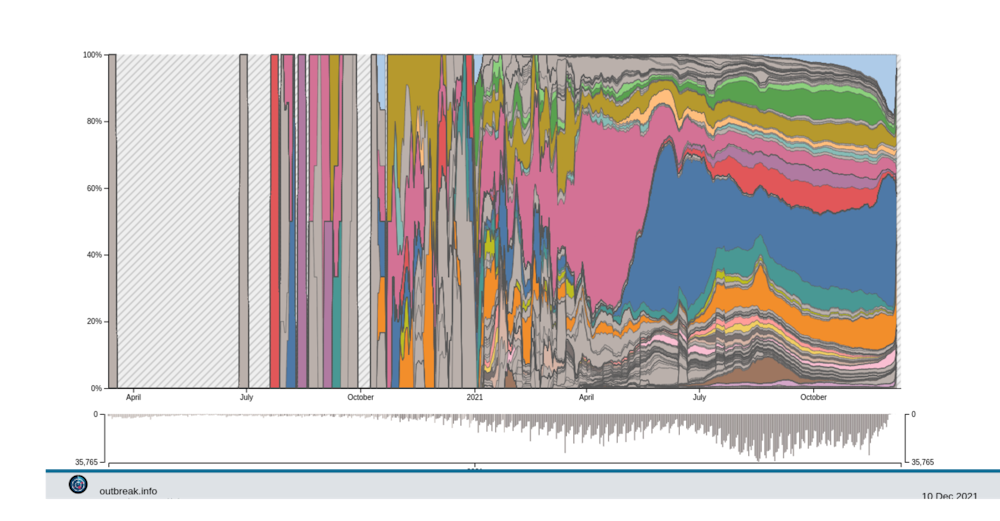
T.C. : Delta peut-il conserver « indéfiniment » cet avantage ?
S.A. : Plus les populations s’immunisent, que ce soit par la vaccination ou, malheureusement, par les infections elles-mêmes, plus l’avantage de Delta s’amenuise. En effet, on sait aujourd’hui que d’autres variants échappent mieux que lui à l’immunité. L’hypothèse la plus commune est donc que Delta soit in fine remplacé par des lignées capables d’infecter des hôtes immunisés. Pour le moment, le variant Beta est celui pour lequel les tests en laboratoire détectent le plus d’échappement immunitaire.
Des expériences avec des protéines virales de synthèse permettent d’anticiper quelles mutations, ou quelles combinaisons de mutations, sont les plus à surveiller.
T.C. : Vous souligniez précédemment qu’ « il faut savoir à quel point le variant Delta est maintenant adapté à nous ». Qu’en est-il ?
S.A. : Avec une contagiosité deux fois plus élevée que les lignées initiales, le variant Delta est sans conteste adapté à court terme à notre espèce. Sur le long terme, c’est moins sûr : cela dépendra de la durée de notre immunité contre l’infection, et des coûts associés à l’échappement immunitaire pour lui. En effet, on sait que certaines mutations permettent au virus d’échapper aux anticorps de patients guéris ou de personnes vaccinées… mais on ne sait pas à quel point ces virus mutés sont contagieux.
M.T.S. : Il est important de garder à l’esprit que la notion d’adaptation, particulièrement dans le cas d’une maladie virale émergente, est relative : le paysage adaptatif évoqué plus tôt est en réalité animé de mouvements comparables à la houle. L’évolution du SARS-CoV-2 illustre bien l’image de la « course aux armements » à laquelle nous nous livrons avec lui : pour le moment nous avons, malgré nous, sélectionné des phénotypes plus contagieux et plus virulents (variants Alpha, Delta). Désormais les regards se portent sur les variants susceptibles de contourner notre deuxième filtre protecteur, que représente l’immunité (post-vaccinale et post-infectieuse).
T.C. : D’où pourrait provenir un nouveau variant ?
M.T.S. : Un variant apparaît comme n’importe quel mutant, au hasard. Chacune des près de 30 000 bases (lettres) du génome du SARS-CoV-2 mute en moyenne tous les 300 000 cycles de division, et une infection peut produire plusieurs milliards de particules virales. Au bout du compte, la grande majorité des individus infectés peut transmettre des virus différents de ceux qui l’ont contaminé. On estime en moyenne que, le long d’une chaîne de transmission, deux mutations se fixent au hasard dans le génome du SARS-CoV-2 chaque mois.
Un mutant particulier est considéré comme un variant s’il présente des changements remarquables d’une ou plusieurs caractéristiques d’intérêt (contagiosité, virulence, échappement immunitaire, symptomatologie ou résistance aux antiviraux). L’émergence d’un variant correspond souvent à bond mutationnel, avec des vitesses évolutives 2 à 4 fois plus élevées.
Au final, chaque infection non évitée est une occasion pour le virus de muter et, potentiellement, d’engendrer un variant. Fort heureusement, ces événements restent très rares, car la majorité des mutations sont délétères.
C’est la population dans laquelle le virus circule qui détermine quelles mutations lui seront, dans ce contexte donné, avantageuses (on dit que les pressions de sélection diffèrent) : si cette population n’est pas du tout immunisée, les lignées les plus contagieuses sont favorisées ; si elle est immunisée, alors les lignées capables d’échapper à cette immunité se propagent davantage.
S.A. : À cela, on peut rajouter le cas des infections chroniques, notamment chez des personnes immunodéprimées. Dans ce cas, le niveau de sélection « intra-patient » se rajoute au niveau de sélection populationnelle. Or il a été montré qu’au cours d’une infection de plusieurs mois, le système immunitaire sélectionne des virus de SARS-CoV-2 ayant des mutations qui ont été retrouvées chez des variants.
En théorie, ce résultat n’est pas automatique et, pour le virus de l’immunodéficience humaine (VIH) par exemple, on pense que l’adaptation intra-patient se fait au détriment de la propagation dans la population. En tout cas, ce résultat fait que la co-circulation du VIH et du SARS-CoV-2 dans des populations non vaccinées comme en Afrique subsaharienne est un enjeu sanitaire majeur, ainsi que cela a déjà été signalé lors de l’évolution du variant Gamma.
T.C. : Que peut-on dire plus précisément de l’origine d’Omicron ?
S.A. : Le nouveau variant Omicron a été identifié en Afrique du Sud, mais il n’est a priori pas apparu là-bas. Ce pays l’a détecté grâce à la qualité de son suivi épidémiologique et génomique. À ce sujet, la réaction de rejet de ce pays de la part de la communauté internationale est problématique, car risque de décourager les efforts de surveillance.
Si l’origine exacte de ce variant est pour le moment inconnue, le plus vraisemblable est qu’il provienne d’une région d’Afrique où le suivi de l’épidémie est limité. En effet, il n’existe quasiment pas de séquences récentes de virus SARS-CoV-2 proches de celle d’Omicron : les analyses de génomes nous indiquent que son ancêtre commun avec les autres variant remonte à mi-2020 ! Cela signifie qu’il proviendrait de lignées qui ont circulé pendant plus d’un an sans être échantillonnées (ce qui est fort possible vu le faible investissement pour le suivi de l’épidémie dans beaucoup de pays d’Afrique).
On pourrait aussi envisager que le virus ait fait un passage par un réservoir animal, car certaines de ses mutations intriguent. On sait que le SARS-CoV-2 peut infecter des mammifères et, dans certains cas, comme dans les élevages de visons, il existe des cas de retours dans la population humaine. Mais pour Omicron, on ne dispose encore que de très peu de données pour explorer cette hypothèse.
T.C. : L’expression de « saut évolutif » a été employée pour certains variants. Qu’est-ce que cela veut dire ?
S.A. : On touche là à un vieux débat en biologie de l’évolution entre les partisans du « gradualisme », que l’on peut faire remonter à Charles Darwin, et ceux des « équilibres ponctués » (les sauts), popularisés par Niles Eldredge et Stephen Jay Gould. La vérité emprunte aux deux et la pandémie actuelle en offre un bon exemple.
On sait que le SARS-CoV-2 accumule naturellement des mutations génétiques à une vitesse assez régulière, et c’est ce qui nous permet de le suivre à la trace dans les études de phylodynamique. Mais l’évolution des variants se traduit aussi par des changements étendus dans le génome, ou « balayages sélectifs » où une mutation bénéfique (et les séquences associées) se fixe : avec d’abord la mutation D614G (au tout début de la pandémie, le 614e acide aminé de la protéine Spike était habituellement un acide aspartique – « D » dans la nomenclature spécialisée ; il est ici remplacé par une glycine, « G », ndlr), puis avec le variant Alpha (les variants Beta et Gamma sont restés minoritaires au niveau mondial) et ensuite le variant Delta.
Donc selon l’échelle à laquelle vous regardez (le mois ou l’année) et les critères que vous utilisez (les mutations neutres ou celles dites phénotypiques affectant les propriétés biologiques telle la contagiosité), vous aurez une vision de continuité ou de sauts.
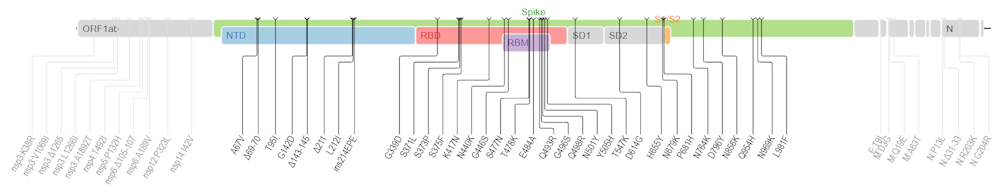
T.C. : Et comment interpréter Omicron, qui présente 53 mutations dont une trentaine sur la seule protéine Spike ?
S.A. : Pour le moment on n’en sait que peu de choses d’Omicron mais, effectivement, son profil mutationnel interpelle. Comme l’analysent l’expert en évolution virale sud-africain Darren Martin et ses collaborateurs, on peut classer ces mutations en trois groupes :
- D’une part, il y a toutes les mutations et délétions qui tournent autour de la mutation N501Y dans la protéine Spike (Δ69-70, K417N, N501Y, H655Y, P681H). Cette dernière a déjà été décrite comme modifiant profondément le paysage adaptatif du virus, c’est-à-dire le champ de ses possibles.
- Ensuite, toujours dans la Spike, il existe un second ensemble de mutations à des positions déjà mutées chez d’autres variants, par exemple en position 484 (N440S, S477I ou E484A). On s’attend à ce qu’elles aient un effet sur le phénotype des infections, par exemple leur contagiosité, leur virulence ou leur capacité d’échappement à la réponse immunitaire.
- Enfin, il existe un ensemble de 14 mutations qui sont très peu présentes dans les lignées circulant, et même quasi absentes des autres sarbecovirus connus (sous-genre de bétacoronavirus regroupant les coronavirus liés au syndrome respiratoire aigu sévère, dont le SARS-CoV-2, ndlr). De plus, individuellement, ces mutations semblent contre-sélectionnées. Leur présence est donc actuellement une énigme, car le variant semble bien adapté à notre espèce. Une possibilité est qu’il existe un effet collectif de ces mutations ou une interaction avec d’autres mutations (telles que la N501Y) selon un phénomène d’épistasie, courant en génétique des populations : même si deux mutations A et B sont délétères isolément, leur présence conjointe peut être avantageuse.
À noter que l’on distingue déjà deux sous-lignées d’Omicron. Elles sont notées BA.1 et BA.2, et n’ont pas toutes les deux les mêmes mutations clés.
T.C. : Quels autres signaux seraient inquiétants, à part ces mutations ?
M.T.S. : Le point le plus frappant est qu’Omicron est associé à une reprise épidémique très forte en Afrique du Sud, juste après la vague de variant Delta. Sur cela, il faut préciser qu’il est très délicat d’appréhender les contextes nationaux. Par exemple, notre équipe déploie beaucoup d’efforts pour comprendre l’épidémie en France. Par conséquent, pour juger de la gravité de la situation épidémique dans un pays, le mieux est de s’en remettre aux épidémiologistes et agences sanitaires de ce pays… Et dans le cas de l’Afrique du Sud, ces spécialistes sont inquiets.
Plusieurs expériences consistant à soumettre le virus à des anticorps issus de personnes vaccinées ou guéries sont en cours. Aucune n’est encore publiée donc il est difficile de tirer des conclusions, d’autant que les résultats préliminaires sont assez divergents entre études – voire au sein de certaines études…
Il faut garder à l’esprit que ces expériences indiquent une tendance générale et ne capturent pas la diversité de la réponse immunitaire. Au final, ce seront les analyses statistiques des études épidémiologiques qui seront les plus utiles. De ce côté, une première étude de terrain suggère une capacité du virus à causer plus de réinfections que les autres lignées. Autrement dit, la croissance rapide d’Omicron pourrait s’expliquer plus par sa capacité à contourner les réponses immunitaires que par son R₀.
T.C. : Quelle peut-être la diffusion réelle d’Omicron au niveau mondial ?
S.A. : Omicron a déjà été détecté à bas bruit dans des dizaines de pays, dont la France. Une telle répartition fait penser à ce qu’on appelle une dynamique « source-puits » en écologie scientifique : on aurait une région du monde où ce virus est majoritaire et un phénomène de dispersion en cours.
Dans plusieurs pays qui ont un bon suivi de leur épidémie, Royaume-Uni et Danemark notamment, on distingue déjà une croissance très rapide des tests cohérents avec ce variant. En effet, comme il comporte une délétion en position 69-70 de la Spike, un des tests de dépistage qui comporte 3 cibles dans le génome rend des tests positifs avec 2 des 3 cibles présentes.
M.T.S. : En France, les données de criblage, qui consistent à chercher des mutations particulières, permettent d’avoir une idée quasiment en temps réel de la propagation d’un groupe de génotypes. L’avantage est que cette technique est moins coûteuse et plus rapide que le séquençage complet des génomes. Seul celui-ci permet toutefois d’identifier avec certitude le variant.
T.C. : A-t-on une idée du nombre de variants (d’intérêt ou préoccupants) qui ont pu apparaître sans être repérés ?
S.A. : Les regards sont évidemment actuellement rivés sur le nouveau variant Omicron, mais il est difficile de dire combien il existe de mutants d’intérêt au niveau planétaire. Il est assez vraisemblable que plusieurs aient émergé sans ensuite percer. En effet, même avec un avantage sélectif, les premiers stades de propagation d’un variant sont gouvernés par le hasard.
M.T.S. : Dans un modèle simpliste, la probabilité d’extinction d’une épidémie est de 1/R₀. En première approximation, avec un R₀ de 3, dans 33 % des cas les chaînes de transmission s’éteignent spontanément. Ainsi, plusieurs introductions du SARS-CoV-2 en France ont pu avoir eu lieu et la quête d’identification d’un cas index est discutable.
C’est aussi pour cela qu’il est possible qu’il y ait eu plusieurs émergences de variants dont les chaînes de transmission se soient éteintes d’elles-mêmes. En effet, les événements de super-propagation (à la faveur de rassemblement sans gestes barrières, etc.) jouent un rôle clé dans la propagation de ce virus, l’essentiel des transmissions étant le fait d’une minorité de cas.
T.C. : Quels sont les autres variants actuels les plus surveillés ?
S.A. : En France, on surveille avec attention la lignée B.1.640, détectée pour la première fois en mars 2021, notamment en République Démocratique du Congo ; elle n’est pas répertoriée comme variant d’intérêt par l’OMS mais semble se propager assez vite. Notre équipe a aussi identifié une circulation de variants Delta portant au moins deux mutations associées à de l’échappement immunitaire dans la protéine Spike (la T95I et la E484Q). Pour le moment, leur circulation reste limitée.
En tout cas, l’émergence du variant Omicron nous rappelle la nécessité d’avoir une vision à long terme pour sortir de cette pandémie. À part le conseil scientifique, peu s’en préoccupent, car cela entre en contradiction avec l’immédiateté du temps politique et médiatique. Si elle est une actrice parmi d’autres, la recherche scientifique a un important rôle à jouer, du développement de traitements jusqu’à l’anticipation de l’évolution virale.
Malheureusement, elle s’inscrit sur le temps long ce qui la rend peu attractive pour les politiques… et elle fait les frais d’une inculture scientifique qui traverse presque tous les milieux de la société française. Avec les récentes suppressions des cours de biologie ou de physique-chimie pour la majorité des lycéennes et lycéens, les croyances aux solutions miraculeuses risquent encore d’empirer.
Samuel Alizon
Directeur de Recherche au CNRS, Institut de recherche pour le développement (IRD)
Mircea T. Sofonea
Maître de conférences en épidémiologie et évolution des maladies infectieuses, laboratoire MIVEGEC, Université de Montpellier
Cet article est republié à partir de The Conversation sous licence Creative Commons. Lire l’article original.







